ABSTRACT
Objective: To evaluate the prevalence of alpha-1 antitrypsin (AAT) variants through SERPINA1 genotyping in patients with non-cystic fibrosis bronchiectasis, and assess their clinical, functional and radiological characteristics. AAT deficiency is underdiagnosed, and an etiology to be considered when evaluating bronchiectasis. Methods: A cross-sectional study was conducted at an outpatient clinic focused on bronchiectasis in a tertiary hospital. Data from patients followed between 2005 and 2023 were collected. Genotyping for AAT was performed. Demographic, clinical, pulmonary function tests, serum AAT levels and chest CT data were analyzed. Results: A total of 136 patients were included, predominantly female (72.1%), with a median age of 56.6 years. The prevalence of SERPINA1 gene mutations was 25.7% (n=35). Among the detected variant genotypes were Pi*MS (15.4%), Pi*MZ (5,1%), Pi*SS (1,5%), Pi*ZZ (1,5%), Pi*MI (0,7%), Pi*SZ (0,7%) and Pi*ZMMalton (0,7%). When comparing patients with and without SERPINA1 mutations, significant differences were observed in AAT serum levels, emphysema type (panlobular) and distribution (diffuse and lower-lobe predominant). No other clinical, microbiological, functional or radiological differences were found, including emphysema presence or absence. Notably, 16 (45.7%) of individuals carrying SERPINA1 mutations exhibited normal serum AAT levels. Conclusions: AAT variants are not uncommon among patients with bronchiectasis. Presence of panlobular, diffuse or lower-lobe predominant emphysema should prompt AATD diagnostic consideration. However, the absence of emphysema does not exclude the diagnosis. Moreover, SERPINA1 variants may occur along with normal AAT serum levels. Clinicians should consider genotyping in patients with normal AAT levels, particularly when bronchiectasis remains unexplained.
Keywords:
Alpha 1-antitrypsin deficiency; Bronchiectasis; Genotype; Mutation; Alleles
INTRODUCTION Alpha-1 antitrypsin deficiency (AATD) is a still underdiagnosed autosomal codominant disorder caused by mutations in the
SERPINA1 gene, resulting in reduced serum levels of alpha-1 antitrypsin (AAT) and increased susceptibility to chronic pulmonary and liver diseases. (1) AAT is the main circulating serine protease inhibitor, and its deficiency promotes a protease–antiprotease imbalance, favoring lung tissue damage to occur.(2) Although AATD is typically associated with emphysema, recent studies have also linked the condition to other lung diseases, such as asthma and non-cystic fibrosis bronchiectasis.(3-5)
Bronchiectasis is a chronic suppurative lung disorder, caused by a heterogenic group of diseases, characterized by permanent bronchial dilation, typically presenting with chronic cough, sputum production, dyspnea and recurrent respiratory infections. Identifying the underlying etiology is critical to guide management and prognosis.(6) Despite thorough evaluation, 24-40% of bronchiectasis cases remain undetermined worldwide.(7,8) Therefore, AATD should be systematically considered as a possible cause of bronchiectasis.(3,6)
Although the prevalence of AATD is unknown in most countries and varies across populations, it is most frequently observed in individuals of European ancestry,(3) with estimates of up to 1 in 5,000 people in Europe.(9) In Brazil, epidemiological data of AATD in general population are lacking.(10) A cross-sectional study involving 926 patients with COPD from five different Brazilian regions found an overall prevalence of 2.8% for AATD.(11) However, to our knowledge, there are no published data specifically addressing the prevalence of AAT variants among patients with bronchiectasis in Brazil.
This study aims to evaluate the prevalence of AAT variants in a population of patients with bronchiectasis using
SERPINA1 gene mutation testing, and to assess their clinical, functional and radiological characteristics compared to those without mutations.
METHODS This was a cross-sectional study, performed at the non-cystic fibrosis bronchiectasis referral center of the Federal University of Paraná, southern Brazil. Between 2005 and 2023, 239 patients were followed at the outpatient clinic. Data were evaluated and collected from July 2022 to December 2023. The present study was approved by the Committee for Ethics in Research on Human Beings under the opinion 5.464.008, and written informed consent was obtained from all participants.
Patients aged 18 years or older with bronchiectasis diagnosed by clinical and tomographic criteria, who had undergone genotyping for AAT variants were included. Patients with insufficient data or a diagnosis of cystic fibrosis were excluded.
Diagnosis and etiology of bronchiectasis were defined according to specific diagnostic criteria.(6,12) For this study, etiology was grouped as follows: post-tuberculosis, post-infectious (other), common variable immunodeficiency, immunoglobulin A deficiency, related to HIV virus infection, other immunodeficiency, related to auto-immune diseases (collagen and inflammatory bowel diseases), primary ciliary dyskinesia, AATD-related, undefined (or idiopathic), and others (involving the diagnosis: Williams-Campbell syndrome, Scimitar syndrome, allergic bronchopulmonary aspergillosis, chronic aspiration).
Clinical, laboratory, and imaging data were retrieved from electronic medical records and hospital information systems and compiled for analysis. Data related to the following were collected:
- Clinical variables: age, sex, race, smoking status, BMI, long-term oxygen therapy, number of exacerbations and hospitalizations in the previous 12 months of follow-up, comorbidities (asthma and prior tuberculosis), and E-FACED bronchiectasis severity score (calculated based on occurrence of severe exacerbation in the previous year, FEV1 % predicted, age, chronic infection by Pseudomonas aeruginosa, radiological extent, and dyspnea by modified Medical Research Council scale).
- Laboratory variables: serum AAT level, SERPINA1 genotype, sputum culture microbiology.
- Pulmonary function: FEV1, FVC, FEV1/FVC ratio, DLCO (absolute and % predicted) and bronchodilator response were evaluated. FEV1 was considered very severely reduced when <30% predicted; severely reduced when 30–49% predicted; moderately reduced when 50–79% predicted and mildly reduced when ≥80% predicted.
- CT scan variables: images were acquired in a multidetector scanner (Aquilion 64; Toshiba Medical Systems, Tokyo, Japan), with a high-resolution protocol (1-mm slice thickness and 0.5-mm increments), and assessed by a chest radiologist. Evaluated parameters included presence, type and distribution of emphysema and bronchiectasis, number of affected lobes, bronchial wall thickening, mucus plugging, tree-in-bud opacities, and prior surgical resections.
AAT measurement and genotypingSerum AAT levels were measured by turbidimetry in peripheral venous blood samples. The reference range for this method was 90–200 mg/dL. By protocol, measurements were performed during clinically stable periods, as AAT is an acute-phase reactant and may be elevated during inflammatory or infectious exacerbations.(3,10)
Serum AAT concentrations were stratified into three categories according to the AATD diagnostic algorithm(3,10) and the internationally recognized threshold for severe deficiency(13): ≥116 mg/dL (normal), 57–115 mg/dL (intermediate levels), and <57 mg/dL (severely reduced levels).
Genotyping of the
SERPINA1 gene was performed, regardless of AAT serum levels. It was used a buccal swab collection kit (ORAcollect OCR-100; DNA Genotek, Inc, Ottawa, Canada), preserved in bacteriostatic stabilizing solution. Samples were shipped at room temperature and analyzed by Progenika Biopharma S.A. (a Grifols company, Derio, Spain). DNA was extracted and amplified by PCR, followed by hybridization with allele-specific probes targeting 14 common variants in exons II, III, and V, using Luminex xMAP technology (Luminex Corp., Austin, TX, USA).
Genetic diagnosis of AAT variants was confirmed when a mutation was detected in at least one allele. (3,10) AATD was considered a definite etiology of bronchiectasis when severe genotypes were found, such as Pi*ZZ, Pi*SZ and Pi*ZM(Malton).(14)
Statistical analysis Exploratory analysis of quantitative variables was performed by calculating means and standard deviations (mean ± SD) or medians and interquartile ranges (IQR), according to data distribution. Categorical variables were expressed as absolute and relative frequencies. Association analysis between nominal, ordinal, or discrete variables were evaluated using the Chi-square test or Fisher’s exact test, when appropriate. For contingency tables with more than two categories and low expected frequencies, the Fisher-Freeman-Halton exact test was used to ensure statistical validity. For continuous outcome variables, normally distributed data were analyzed using one-way ANOVA, while the Kruskal–Wallis and Mann–Whitney U tests were used for non-normally distributed variables. All statistical analyses were performed with the IBM SPSS Statistics software package, version 29.0 (IBM Corporation, Armonk, NY, USA). A two-tailed p-value < 0.05 was considered statistically significant.
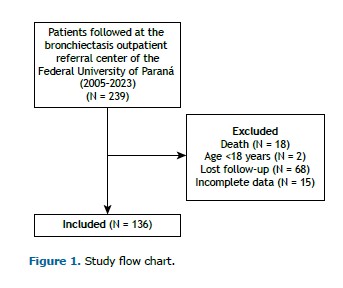
RESULTS Of 239 patients with non-cystic fibrosis bronchiectasis followed at the outpatient referral center between 2005 and 2023, 136 met the eligibility criteria and were included in the study (Figure 1).
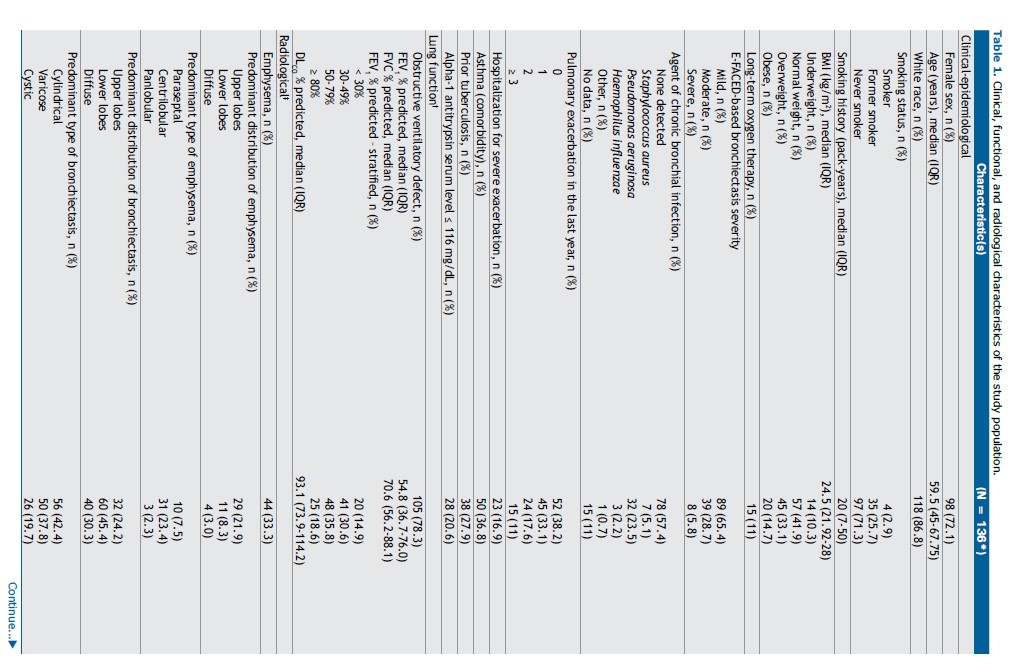
Study population characteristics can be seen in Table 1. The majority were female (72.1%) and White (86.8%). Ages ranged from 18 to 85 years, with a median of 59.5 years (IQR: 45.0–67.8). Only 10.3% were underweighted. Most patients were never smokers (71.3%), and from those who were current or former smokers (28.6%), median smoking history was 20 pack-years (IQR: 7-50).
Chronic hypoxemic respiratory failure requiring long-term oxygen therapy was present in 15 patients (11%). In terms of clinical outcomes, 28.6% had two or more exacerbations in the last year requiring antibiotic treatment, and 16.9% required hospitalization due to severe exacerbation. According to the E-FACED severity score, 34.5% had severe or moderate disease.
Microbiological analysis of spontaneous sputum cultures showed that 57.4% of patients did not have chronic bronchial infection. Among those with infection, Pseudomonas aeruginosa was the most prevalent pathogen (23.5%).
Pulmonary function tests revealed obstructive ventilatory disorder in 78.3% of patients. Most patients had moderate (35.8%) and severe (30.6%) reduction in forced expiratory volume in one second (FEV1).
Considering chest CT, emphysema was identified in 33.3% of patients. When present, it predominantly involved the upper lobes, and centrilobular emphysema was the most frequent pattern. Bronchiectasis was predominantly located in the lower lobes (45.4%) and most frequently cylindrical (42.4%). Extensive lobar involvement (all lobes) was seen in 48.5% of cases. Bronchial wall thickening (68.2%), mucus plugging (70.4%), and centrilobular tree-in-bud opacities (65.9%) were also common.
Regarding etiology (Table 2), post-tuberculosis bronchiectasis was the most common identified cause (21.3%), followed by post-infectious bronchiectasis due to other pathogens (16.9%). Only 2.9% were attributed to AATD.(15) The etiology remained undefined in 40.4% of cases, despite extensive investigation.
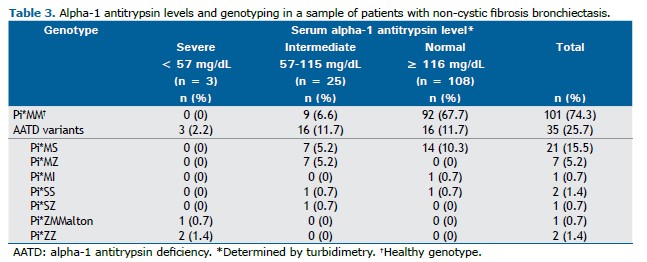
AAT levels and genotyping Twenty-eight individuals with bronchiectasis (20.6%) had low serum levels of AAT (≤116mg/dL), including 2.2% with severe deficiency (<57mg/dL) and 18.4% with intermediate concentrations (57-115mg/dL). Furthermore, 35 (25.7%) had at least one
SERPINA1 mutation identified (Table 3).
Presence of
SERPINA1 mutations was found in 100% of patients with severely reduced AAT levels. However, pathogenic variants were also identified in individuals with serum levels ≥57 mg/dL, including some above normal threshold.(3,10)
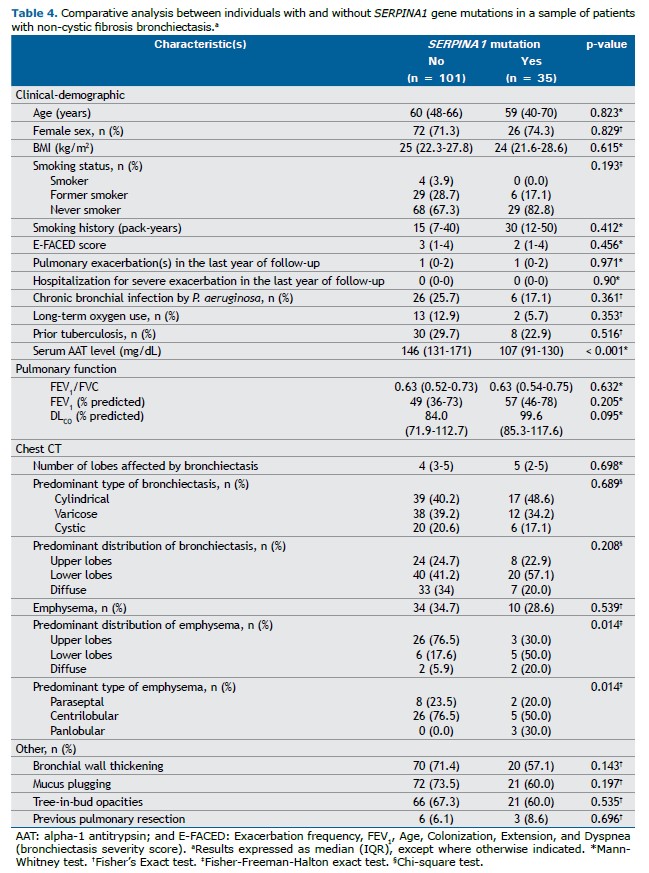
Comparison between patients with and without SERPINA1 gene mutationsWhen the cases were stratified by the presence or absence of
SERPINA1 gene mutations (Table 4), significant differences were found for AAT serum levels, emphysema distribution and emphysema type. The median serum AAT level was 107 mg/dL (IQR: 91–130) among mutation carriers, compared to 146 mg/dL (IQR: 131–171) in non-carriers (p < 0.001). Although the overall frequency of emphysema was similar between groups, among patients with AAT mutations, emphysema was more frequently located in lower lobes (57.1%) or diffusely distributed (20%). Panlobular emphysema was observed exclusively in patients with
SERPINA1 mutations (3 cases, 8.6%).
No statistically significant differences were observed in other demographic, clinical, microbiological, functional, or radiological features.
DISCUSSION To our knowledge, this is the first study to report the prevalence of AAT variants in a population of patients with non-cystic fibrosis bronchiectasis in Brazil, in which
SERPINA1 gene mutations were identified in 25.7% of the individuals. It is important to highlight that AATD is a genetically determined condition more commonly observed in Caucasian populations. Given Brazil’s racial admixture and continental dimensions, along with historical differences in regional immigration patterns, it is plausible that the prevalence of AAT variants may vary across regions of the country.
In Brazil, the prevalence of AAT variants was previously assessed only in a cohort of 926 individuals with COPD from five different regions.(11) In that study, genetic analysis was performed only in the individuals with reduced serum AAT levels identified during initial screening. In contrast, the present study conducted genotyping in the entire sample of patients with non-cystic fibrosis bronchiectasis, which may partially explain the higher prevalence of
SERPINA1 mutations observed (25.7% vs. 2.8%).
When compared to studies focusing on bronchiectasis populations, our findings also revealed a higher frequency of
SERPINA1 mutations. In Italy, a study that investigated the prevalence of AAT variants by genotyping in a population with bronchiectasis found a frequency of 7.7%.(15) In the United Kingdom, AATD prevalence among 1,600 patients with bronchiectasis was 0.5%, through initial evaluation of serum levels, followed by phenotyping when levels were low.(16) In France, AATD diagnosis based directly on phenotyping method in a population of 202 patients with bronchiectasis showed a frequency of 18.81%.(17)
When comparing the two groups—those with and without
SERPINA1 mutations—significant differences were found only in serum AAT levels, predominant pattern of emphysema distribution (lower lobes or diffuse), and emphysema type (panlobular). These findings are consistent with previous reports suggesting that AATD should be suspected in patients with bronchiectasis who exhibit low serum AAT levels and imaging features of basal or panlobular emphysema. (18) However, it is noteworthy that emphysema was present in only 26.8% of mutation carriers in this sample, and only 30% of these cases had panlobular emphysema. Both cases carrying Pi*ZZ genotype did not have emphysema. Therefore, the absence of emphysema—especially of the panlobular or basal type—should not preclude investigation for AATD. This is further supported by data from the European Alpha-1 Research Collaboration (EARCO) international registry, in which 9.1% of Pi*ZZ carriers presented with bronchiectasis in the absence of radiological emphysema.(4)
Although serum AAT levels were lower in the mutation group, normal values did not exclude the presence of AAT variants. In fact, 16 (45.7%) of the 35 patients with
SERPINA1 gene mutations had serum concentrations above the normal threshold.(3) Of note, 14 of these 16 patients carried the Pi*MS genotype. The remaining two patients had the following genotypes: Pi*MI and Pi*SS. Although the Pi*MS genotype alone is not historically considered a significant risk factor for the development of pulmonary disease,(14,19) and therefore cannot be considered a definitive etiological factor for bronchiectasis, it is conceivable that cumulative exposure to other risk factors—such as smoking, occupational exposures, and recurrent respiratory infections—might modulate this risk. Interestingly in the present study, one patient with the Pi*MS genotype and undefined etiology for bronchiectasis presented with panlobular emphysema affecting a lower lobe, despite never having smoked.
A rare genotype identified in our cohort was Pi*MI, involving the I allele, which is classified as a variant of uncertain clinical significance.(3) This was in a young adult, never-smoker, with a serum AAT level of 141 mg/dL, who presented with bronchiectasis of indeterminate etiology, along with coexistent upper lobe emphysema. Pulmonary function was markedly impaired (FEV1 = 38% of predicted), and the patient also had evidence of chronic liver disease of unknown cause. This case raises the possibility that certain rare
SERPINA1 variants, although not classically associated with severe deficiency, may contribute to complex phenotypes involving both pulmonary and extrapulmonary manifestations. Notably, despite serum AAT levels within the normal range, the functional activity of the molecule may be impaired, as described for certain variants that produce structurally unstable or dysfunctional proteins.(19,20)
Another point to be considered is that chronic inflammation, a hallmark of bronchiectasis,(21) may increase AAT concentrations due to its role as an acute-phase reactant.(10,20) Thus, diagnosing AATD solely on the basis of the initial serum AAT level, without subsequent confirmation using complementary diagnostic tools (such as genotyping, phenotyping, or gene sequencing), may lead to underdiagnosis in this population.
Regarding microbiology findings, data from the U.S. Bronchiectasis Registry previously demonstrated increased nontuberculous mycobacterial (NTM) infections in patients with AATD.(14) This may be explained by at least two mechanisms: AAT enhances macrophage-mediated clearance of intracellular mycobacteria through increased phagolysosomal fusion and autophagy,(22) and it potentiates TNF-α activity. Thus, low serum AAT levels may reduce TNF-α–mediated immune responses, increasing susceptibility to mycobacterial infections.(23) Although this pathophysiological rationale exists, no significant differences were observed in AAT variant carriers in this study.
In the present study, no other clinical, laboratory, or radiological variables differed significantly between patients with and without
SERPINA1 variants. Thus, apart from low serum AAT levels, diffuse or basal emphysema distribution, and panlobular emphysema, no consistent pattern could be identified to suggest the presence of
SERPINA1 mutations in individuals with bronchiectasis. Importantly, these findings should not be used as exclusive criteria to guide genetic investigation, since, as previously discussed, a substantial proportion of individuals carrying
SERPINA1 mutations may present with normal serum AAT levels and without radiological evidence of emphysema.
This study has limitations. This is a cross-sectional analysis based on consecutive patients evaluated in a referral center over a defined period. As such, no formal sample size calculation was performed. The single-center nature of the study limits the generalizability of the findings. Furthermore, the rarity of bronchiectasis associated with AATD led to a relatively small number of cases, potentially reducing the statistical power for detecting significant associations. Additionally, serum AAT levels were measured using turbidimetry, which, although the method available at our center, is known to be less sensitive than nephelometry—the technique preferably indicated by consensus.(3) This lower sensitivity is particularly relevant for detecting subtle differences at lower AAT concentrations. Despite these limitations, our study provides a comprehensive clinical, microbiological, radiological, and genetic characterization of a significant population of patients with non-cystic fibrosis bronchiectasis in a referral center. Although numerous studies have explored bronchiectasis among individuals with AATD, investigations focusing on screening for AAT variants in populations with primary bronchiectasis, especially at referral centers, remain scarce.
In conclusion, our findings suggest that AAT variants are not uncommon among patients with bronchiectasis. Clinicians should consider genotyping or further diagnostic evaluation even in patients with normal AAT levels, particularly when bronchiectasis remains unexplained. Although the identification of a
SERPINA1 mutation alone may not always define the etiology of bronchiectasis, it should be considered an additional risk factor that may act synergistically with other environmental or infectious exposures to influence disease expression.
ACKNOWLEDGMENTS The authors would like to thank all of the patients living with non-cystic fibrosis bronchiectasis who participated in and contributed to this study. We are also grateful to Progenika Biopharma S.A. (Derio, Spain), a Grifols Company, for their support in the genetic analysis.
AUTHOR CONTRIBUTIONS CSS, MGMC, and KMS: study conception and design; drafting and reviewing the manuscript. CAL: drafting and reviewing the manuscript. All authors read and approved the final version of the manuscript.
CONFLICTS OF INTEREST None declared.
REFERENCES 1. Miravitlles M, Dirksen A, Ferrarotti I, Koblizek V, Lange P, Mahadeva R, et al. European Respiratory Society statement: Diagnosis and treatment of pulmonary disease in Alfa1-antitrypsin deficiency. Eur Respir J. 2017;50(5):1700610. https://doi.org/10.1183/13993003.00610-2017
2. Stockley RA, Parr DG. Antitrypsin deficiency; still more to learn about the lung after 60 years. ERJ Open Res. 2024;10(4):00139-2024. https://doi.org/10.1183/23120541.00139-2024
3. Feitosa PHR, Castellano MVCO, Costa CHD, Cardoso ADRO, Pereira LFF, Fernandes FLA, et al. Recommendations for the diagnosis and treatment of alpha-1 antitrypsin deficiency. J Bras Pneumol. 2024;50(5): e20240235. https://doi.org/10.36416/1806-3756/e20240235
4. Stockley RA, Pye A, De Soyza J, Turner AM, Miravitlles M. The prevalence of bronchiectasis in patients with alpha-1 antitrypsin deficiency: initial report of EARCO. Orphanet J Rare Dis. 2023;18(1):1-8. https://doi.org/10.1186/s13023-023-02830-2
5. Soyza J De, Ellis BP, Newnham M, Rickard L, Turner MAM. Bronchiectasis Occurs Independently of Chronic Obstructive Pulmonary Disease in Alpha-1 Antitrypsin Deficiency. Chronic Obstr Pulm Dis. 2024;11(5):507-514. https://doi.org/10.15326/jcopdf.2024.0526
6. Pereira MC, Athanazio RA, Dalcin PTR, Figueiredo MRF, Gomes M, Freitas CG, et al. Brazilian consensus on non-cystic fibrosis bronchiectasis. J Bras Pneumol. 2019;45(4):e20190122. https://doi.org/10.1590/1806-3713/e20190122
7. Lonni S, Chalmers JD, Goeminne PC, Mcdonnell MJ, Dimakou K, De Soyza A, et al. Etiology of Non-Cystic Fibrosis Bronchiectasis in Adults and Its Correlation to Disease Severity. Ann Am Thorac Soc. 2015;12(12):1764-70. https://doi.org/10.1513/AnnalsATS.201507-472OC
8. Olveira C, Padilla A, Martínez-García MÁ, De La Rosa D, Girón RM, Vendrell M, et al. Etiology of Bronchiectasis in a Cohort of 2047 Patients. An Analysis of the Spanish Historical Bronchiectasis Registry. Arch Bronconeumol. 2017;53(7):366-374. https://doi.org/10.1016/j.arbres.2016.12.003
9. Orphanet Report Series [homepage on the Internet]. Paris: Orphanet; c2024. Prevalence and incidence of rare diseases: Bibliographic data. [Adobe Acrobat doc. 194p.].. Available from: http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf
10. Jardim JR, Casas-Maldonado F, Fernandes FLA, Castellano MVCO, Torres-Durán M, Miravitlles M. Update on and future perspectives for the diagnosis of alpha-1 antitrypsin deficiency in Brazil. J Bras Pneumol. 2021;47(3):e20200380. https://doi.org/10.36416/1806-3756/e20200380
11. Russo R, Zillmer LR, Nascimento OA, Manzano B, Ivanaga IT, Fritscher L, et al. Prevalence of alpha-1 antitrypsin deficiency and allele frequency in patients with COPD in Brazil. J Bras Pneumol. 2016;42(5):311-6. https://doi.org/10.1590/S1806-37562015000000180
12. Araújo D, Shteinberg M, Aliberti S, Goeminne PC, Hill AT, Fardon T, et al. Standardised classification of the aetiology of bronchiectasis using an objective algorithm. Eur Respir J. 2017;50(6):3-6. https://doi.org/10.1183/13993003.01289-2017
13. Miravitlles M, Anzueto A, Barrecheguren M. Nine controversial questions about augmentation therapy for alpha-1 antitrypsin deficiency: a viewpoint. Eur Respir Rev. 2023;32(170):1-12. https://doi.org/10.1183/16000617.0170-2023
14. Eden E, Choate R, Barker A, Addrizzo-Harris D, Aksamit TR, Daley CL, et al. The Clinical Features of Bronchiectasis Associated with Alpha-1 Antitrypsin Deficiency, Common Variable Immunodeficiency and Primary Ciliary Dyskinesia--Results from the U.S. Bronchiectasis Research Registry. Chronic Obstr Pulm Dis. 2019;6(2):145-153. https://doi.org/10.15326/jcopdf.6.2.2018.0156
15. Aliberti S, Gramegna A, Seia M, Malvestiti F, Mantero M, Sotgiu G, et al. Alpha1-Antitrypsin Inherited Variants in Patients With Bronchiectasis. Arch Bronconeumol. 2023;59(6):401-2. https://doi.org/10.1016/j.arbres.2023.01.004
16. Carreto L, Morrison M, Donovan J, Finch S, Tan GL, Fardon T, et al. Utility of routine screening for alpha-1 antitrypsin deficiency in patients with bronchiectasis. Thorax. 2020;75:592-3. https://doi.org/10.1136/thoraxjnl-2019-214195
17. Cuvelier A, Muir JF, Hellot MF, Benhamou D, Martin JP, Bénichou J, et al. Distribution of α1-antitrypsin alleles in patients with bronchiectasis. Chest. 2000;117(2):415-9. https://doi.org/10.1378/chest.117.2.415
18. Hill AT, Sullivan AL, Chalmers JD, De Soyza A, Elborn S, Floto RA, et al. BTS Guidelines for Bronchiectasis 2018. Thorax. 2019;74(1):1-69. https://doi.org/10.1136/thoraxjnl-2018-212468
19. Stoller JK, Hupertz V, Aboussouan LS. Alpha-1 Antitrypsin Deficiency. 2006 Oct 27 [updated 2023 Jun 1]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 20301692.
20. Franciosi AN, Fraughen D, Carroll TP, McElvaney NG. Alpha-1 antitrypsin deficiency: clarifying the role of the putative protective threshold. Eur Respir J. 2022;59(2):1-12. https://doi.org/10.1183/13993003.01410-2021
21. Long MB, Chotirmall SH, Shteinberg M, Chalmers JD. Review Rethinking bronchiectasis as an inflammatory disease. Lancet Respir. 2024;2600(24):1-14. https://doi.org/10.1016/S2213-2600(24)00176-0
22. Bai X, Bai A, Honda JR, Eichstaedt C, Musheyev A, Feng Z, et al. Alpha-1-Antitrypsin Enhances Primary Human Macrophage Immunity against Non-tuberculous Mycobacteria. Front Immunol. 2019;10:1417. https://doi.org/10.3389/fimmu.2019.01417
23. De Smet S, Dierick J, Steyaert S, Schurgers M, Van Steenkiste C, Loof S. Alfa-1-antitrypsin deficiency: a predisposing factor leading to invasive infec-tions? Infect Dis (Auckl). 2020;52(2):130-4. https://doi.org/10.1080/23744235.2019.1690163






 English PDF
English PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket






